












Синдром Пирсона – это очень редкое генетическое заболевание, которое проявляется еще в младенчестве и в большинстве случаев ведет к раннему летальному исходу.
История открытия
Другое название синдрома Пирсона – врожденная сидеробластная анемия с экзокринной недостаточностью поджелудочной железы. Болезнь названа в честь ученого, впервые ее описавшего в 1979 году – Н. А. Пирсона. Синдром был распознан благодаря длительным наблюдениям за четырьмя детьми со схожими симптомами: у них наблюдалась сидеробластная анемия, которая не поддавалась стандартному лечению, недостаточность экзокринной функции поджелудочной железы и патология клеток костного мозга.
Сначала детям ставили другой диагноз – синдром Швахмана (врожденная гипоплазия поджелудочной железы). Но после исследования крови и костного мозга были выявлены явные отличия, что и дало повод выделить Пирсона синдром в отдельную категорию.
Причины возникновения болезни
Исследование причин заболевания заняло около десяти лет. Врачи-генетики сумели найти генетический дефект, который ведет к делению и дупликации митохондриальной ДНК.
Хотя болезнь и является генетической, обычно мутация появляется спонтанно, и больной малыш рождается у абсолютно здоровых родителей. Иногда отмечают зависимость между наличием офтальмопатии у матери и развитием синдрома Пирсона у ее ребенка.
Дефекты ДНК удается выявить в костном мозге, ациноцитах поджелудочной железы, а также в органах, которые не являются главными мишенями болезни – почках, сердечной мышце, гепатоцитах. С другой стороны, у некоторых пациентов при наличии типичной клинической и лабораторной картины так и не удается зарегистрировать изменения в митохондриальной ДНК.
У больных детей происходит накопление железа в печени, склероз клубочков почек, образование кист. В некоторых случаях развивается фиброз миокарда, что приводит к сердечной недостаточности.
Поджелудочная железа выделяет недостаточное количество липазы, амилазы и бикарбонатов у всех пациентов с болезнью Пирсона. Синдром проявляется атрофией ткани железы и ее последующим фиброзом.
Методы диагностики
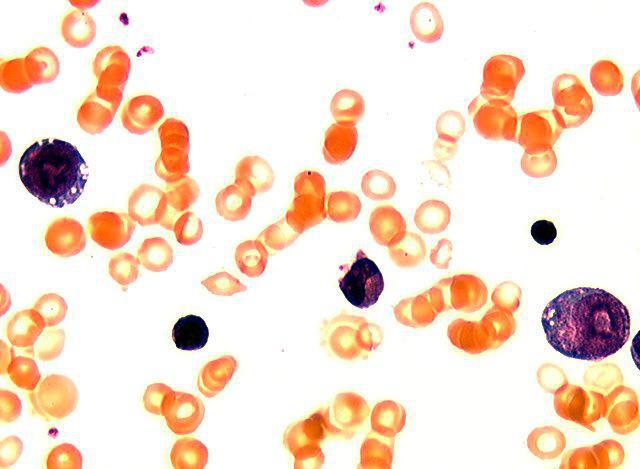
С уверенностью поставить диагноз могут только врачи-генетики после исследования митохондриальной ДНК. Также важную роль играет обычный анализ периферической крови: выявляют макроцитарную анемию тяжелой степени, нейтропению и тромбоцитопению. Примечательным является отсутствие эффекта от лечения анемии "Цианкобаламином" и препаратами железа.
Благодаря пункции костного мозга можно увидеть уменьшение общего количества клеток, наличие вакуолей в эритробластах и появление кольцевидных сидеробластов.

Симптомы болезни
Уже с первых дней жизни ребенка можно заподозрить синдром Пирсона. Симптомы болезни дебютируют у младенцев в виде злокачественной анемии и инсулинзависимого сахарного диабета. Наблюдаются бледность кожи, сонливость, вялость, диарея, периодическая рвота, ребенок плохо прибавляет в весе. Пища почти не усваивается, характерна стеаторея. Возникают симптомы сахарного диабета, повышается уровень глюкозы в крови, и появляется склонность к ацидозу. Возможно развитие печеночной, почечной и сердечной недостаточности.
Иногда кроме анемии возникает панцитопения (дефицит не только эритроцитов, но и тромбоцитов и лейкоцитов), что будет проявляться склонностью к кровотечениям и частому присоединению инфекций.

Лечение и прогноз
К сожалению, врачи до сих пор не знают, как победить синдром Пирсона. Лечение его неспецифическое и дает лишь кратковременные результаты.
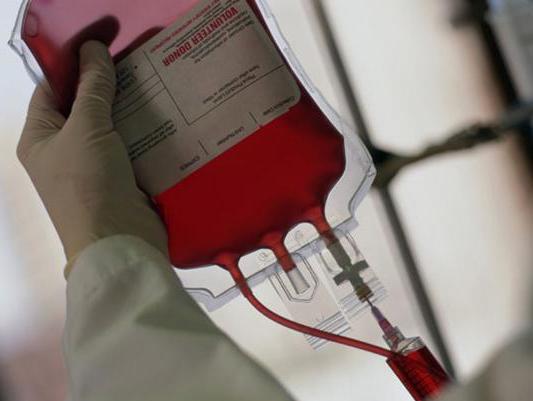
Анемия не поддается стандартной терапии и требует частого переливания крови. Для улучшения функции поджелудочной железы назначают прием ферментов, а для коррекции метаболических нарушений – инфузионную терапию. В редких случаях проводят трансплантацию костного мозга.
Пирсона синдром имеет неблагоприятный прогноз: дети отстают в физическом развитии, большинство погибает до двух лет. В единичных случаях пациенты живут дольше благодаря эффективной поддерживающей терапии, однако в более старшем возрасте болезнь приводит к мышечной атрофии, характерной для синдрома Кернса-Сейра.
Тяжесть течения болезни во многом зависит от степени поражения ДНК.











